> Perl is remarkably good for slicing, dicing, twisting, wringing, smoothing, summarizing and otherwise mangling text. … Perl's powerful regular expression matching and string manipulation operators simplify this job in a way unequalled by any other modern language.
Indeed! I still think in terms of PCRE (Perl-compatible regular expressions), and I love that Perl makes regexes a first-class citizen.
> Although the biological sciences do involve a good deal of numeric analysis now, most of the primary data is still text: clone names, annotations, comments, bibliographic references. Even DNA sequences are textlike. Interconverting incompatible data formats is a matter of text mangling combined with some creative guesswork.
This is still true! One common format (arguably the most common format) for sending around bits of sequenced DNA is the FASTQ format (https://en.wikipedia.org/wiki/FASTQ_format). FASTQ files are (ASCII) plain text, making them really easy to parse. Of course one byte per letter of DNA is wasteful, so FASTQ files are commonly exchanged GZIP-compressed, with the .fastq.gz extension. Many platforms & tools read in or write out .fastq.gz automatically, saving you the (de)compression step.
I went down a rabbit hole looking at how they distributed the original sequence for Covid-19 out of Wuhan. Pretty amazing to go back to January 2020 and see the conversations unrolling out in the open.[0] Also, here's the original sequence in FASTA format.[1] It's incredible to think that you can just email these files around, they're just text meant to be parsed by Perl!
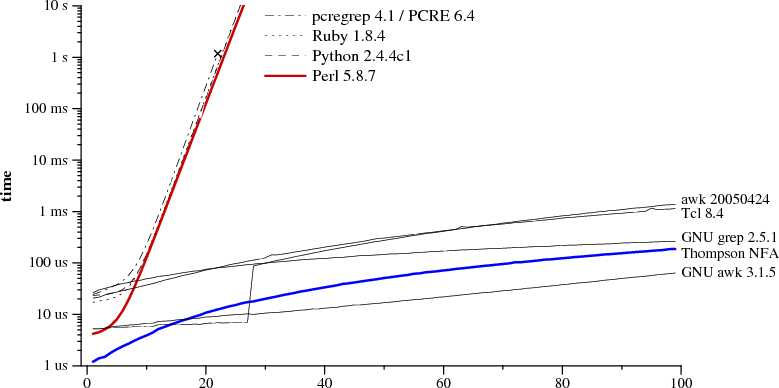
I think at this point something as expressive as PCRE should be table stakes for any language aspiring to be used for text processing. It's so successful that GNU grep added support:
-P, --perl-regexp
Interpret I<PATTERNS> as Perl-compatible regular expressions (PCREs). This option is experimental when combined with the -z (--null-data) option, and grep -P may warn of unimplemented features.
PCRE patterns are inherently unmaintainable; they're not (naturally) compositional or testable. They also cause unpleasant surprises due to accidental Turing completeness (indeed that's one of the more common causes of production outages that I've seen). IMO it's past time for newer languages to offer better alternatives, e.g. really good parser combinator support in the standard library.
Still, when you have a well-defined input and you can treat data as a flat string (no hierarchy), PCRE is probably the best choice you have - and certainly the fastest one.
The biggest surprise for many, myself included, around genomic software that arose after the Human Genome Project was that the useful things people wanted to do with sequence data didn’t need need the expressiveness provided by PCRE, et. al. String algorithms have a minor role in day-to-day genomics.
As mentioned in other comments, sequence analysis is probabilistic, so "matchers" instead tend to be statistical models, like HMMs. There is a rich relationship between statistical models like HMMs and parsing theory.
Really? String algorithms (in the stringology / compressed data structures sense) are the foundation of virtually every operation in genomics that interacts with raw data. Have you ever aligned a sequence?
Regexes are not important. But they are a tiny bit of string algorithms.
Not the op. Yes, I've done some work on this, then tested it against some of the software used in various environments for this kind of work and more than once spotted alternative, more efficient alignments. The practical upshot of that is that I ended up wondering if there is ever a serious bug found in such a piece of software if it shouldn't automatically cause all papers that used the software for their work to be at a minimum flagged for an additional round of review as well as potentially from being disqualified.
What also struck me is that the people using this software treat it like a black box, they have absolutely no way of verifying that what it did it did right.
You’re making a good point that’s usually ignored outside of genomics. Inside genomics, alignment is treated, correctly, as a probabilistic rather than deterministic process (i.e., an alignment is not “right” or “wrong”) and many choose to consider multiple alignments.
That works as long as you don't try to do things like phylogenetic trees, the ordering becomes so critical that even one swap can make things look like the order was the reverse. Of course you should never rely on just the one datum but the temptation to do so and to treat the software as correct is large due to the pressure to publish rather than to hold back and wait until there are multiple pieces of evidence.
There are probably serious bugs in all sequence alignment software, but it's unclear how much it does matter. Downstream analysis must assume that the alignments contain all kinds of known and unknown errors anyway. The sequence alignment problem itself is so ill-defined and there are so many sources of bias and errors, from data to algorithms, from code to reference sequences, from instruments to sample preparation, and including your definition of truth, that you often can't say confidently whether the alignment is correct. The scale of the data is often also big enough that you have to make deliberate trade-offs between costs and correctness.
I have! I even regularly contribute to a popular alignment application! Regardless, you’re absolutely correct and I should’ve chosen my words more carefully. Especially about the relationship to information theory. I meant “regular expressions, LL, LR, LALR, etc.”
In modern genomics, you are seeing more and more use of BAMs (Binary versions of SAM files which themselves are Sequence Alignment Files) even for unaligned data that FASTQ was normally used for. Not only are they smaller in size, but they themselves have a compressed format called CRAM which can use lossy or lossless compression depending on the use case.
Interestingly almost all of the petabyte scale and beyond processing of genomes (whole or exome) is done on the JVM as the library and toolkit ecosystem is extremely mature and it is significantly more performant than just scripting things in Perl. Having access to the big data ecosystem that runs on the JVM is also another reason why languages like Java and Scala are found in the high performance areas of genomics.
> Perl programs are easy to write and fast to develop. The interpreter doesn't require you to declare all your function prototypes and data types in advance, new variables spring into existence as needed, calls to undefined functions only cause an error when the function is needed. The debugger works well with Emacs and allows a comfortable interactive style of development.
I think each and every language that could undo this second "billion dollar mistake", did ("strict mode").
The point is, like PHP and FORTRAN it got millions of people programming who otherwise wouldn't have -- precisely because of its loosey-goosey philosophy (and lack of default strictness). And because it could be used to do some seriously powerful stuff, and get it out the door much more quickly than in the C/C++ world (arguably its only real competitor at the time).
Of course over time, these same people learned to program better, and its looseness and general wackiness grew into a liability.
But the important point here is: language design decisions (just like product decisions) aren't so much intrinsically right or wrong; but right or wrong at certain times.
In its heyday, Perl was, for many people, definitely the right way to go.
{kind=link}
> Perl is remarkably good for slicing, dicing, twisting, wringing, smoothing, summarizing and otherwise mangling text. … Perl's powerful regular expression matching and string manipulation operators simplify this job in a way unequalled by any other modern language.
Indeed! I still think in terms of PCRE (Perl-compatible regular expressions), and I love that Perl makes regexes a first-class citizen.
> Although the biological sciences do involve a good deal of numeric analysis now, most of the primary data is still text: clone names, annotations, comments, bibliographic references. Even DNA sequences are textlike. Interconverting incompatible data formats is a matter of text mangling combined with some creative guesswork.
This is still true! One common format (arguably the most common format) for sending around bits of sequenced DNA is the FASTQ format (https://en.wikipedia.org/wiki/FASTQ_format). FASTQ files are (ASCII) plain text, making them really easy to parse. Of course one byte per letter of DNA is wasteful, so FASTQ files are commonly exchanged GZIP-compressed, with the .fastq.gz extension. Many platforms & tools read in or write out .fastq.gz automatically, saving you the (de)compression step.